An important update is available for FreeStyle LibreLinkØ. Check here for more information.
24 Oct 2019

You can find the original article and more like it on the Takara BioView Blog
Over the past several years, the genome editing field has experienced nothing short of a revolution. Recently, this has largely been driven by the development and refinement of CRISPR/Cas9 technology. By introducing double-strand breaks (DSBs) at targeted locations in the genome and leveraging endogenous repair processes, CRISPR/Cas9 provides a simple method for engineering site-specific genomic modifications. However, despite these rapid advances, this method is still imperfect—and often unpredictable. Errors in DSB repair occasionally introduce insertions or deletions, which may (depending on the desired outcome) necessitate additional screening and a trial-and-error approach to designing guide RNAs.
But why guess at something when you can predict it? While we've written previously about our own tips to optimize gene editing experiments, we'd like to turn the spotlight to the Shendure lab at the University of Washington for their recent article (Chen et al. 2019) that not only showed how repair errors are influenced by the surrounding DNA sequence, but also gave us a way to predict them.
To determine whether gene editing outcomes exhibit a predictable pattern, researchers first generated single guide RNAs (sgRNAs) for 12,917 targets, which were packaged into lentiviruses and used to infect Cas9-expressing HEK293T cells. Targeted genomic regions were amplified by PCR, incorporating unique molecular identifiers (UMIs) to allow the linking of a Cas9-mediated edit to the cell of origin.
Sequencing of these amplicons and subsequent data filtering resulted in 1.16 million reads that represented 6,872 unique targets, with editing outcomes divided into three categories: unedited (wild-type), deletion, or insertion. A minority of these outcomes (9.8% of UMIs) corresponded to unedited alleles, while 63.6% and 31.5% of edited alleles corresponded to deletions and insertions, respectively. Insertions were overwhelmingly comprised of 1-bp events (~75.3%), while large events (>25-bp) were much rarer (~1.5%).
Insertion events seemed to be determined by the sequence surrounding the DSB: 85% of 1-bp insertions appeared to be templated by the 17th nucleotide upstream of the cleavage site. Similarly, for 2-bp insertions, 41% appeared to be templated by the 16th and 17th nucleotides upstream of the cleavage site. Interestingly, insertions were associated with an A or T at the 17th nucleotide, while C and G tended to be associated with deletions.
Over 75% of the observed deletion events were mediated by microhomologies (MHs) 1–10 bp in size, with 94.6% of MHs falling between 1 and 4 bp in size. To determine whether these MHs could be used to predict editing outcomes, researchers reused their earlier strategy to generate and test a library of 1,000 targets and sgRNAs that contained MHs of varying sizes (2, 4, or 6 bp). While putative MH-mediated editing outcomes were more readily predicted compared to a random sequence, there was very little change in the relative number of deletions (82% in targeted MHs, 76% in random sequences).
Based on these findings, the researchers then developed a model, termed Lindel, to predict shorter (<30-bp) deletions and 1- to 2-bp insertion events (comprising the vast majority of insertions). After training this model on their data, they compared Lindel's performance to two other models that had been developed by other groups: ForeCasT (Allen, 2019) and inDelphi (Shen, 2018). Performance was determined based on mean squared error.
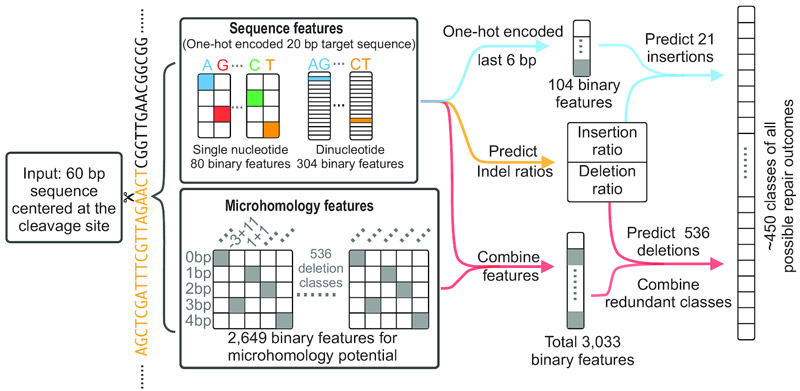
Overview of the underlying methodology of the Lindel target prediction model. Figure by Chen et al. 2019, used under CC BY 4.0.
Perhaps unsurprisingly, Lindel performed best on the researcher's data set (followed by inDelphi and ForeCasT), while ForeCasT performed best on its own data set (followed by Lindel and inDelphi). The researchers then aggregated these two data sets and used the combined data set to re-train Lindel. Following this second training, Lindel had the best performance on both the researcher's data set and the ForeCasT data set—though the researchers noted that most of the prediction errors were primarily due to small deletions and 1-bp insertions, indicating further work will need to be done to predict these events.
The insights gleaned by the Shendure lab will allow researchers a greater measure of predictability in genome engineering experiments. In particular, the Lindel webtool they've provided will narrow down the number of sgRNAs researchers will need to evaluate for their desired editing outcomes, enabling more streamlined and efficient genome editing workflows.
Their tool allows you to predict your experimental outcome—and our kits make those outcomes a reality. Get in touch now to learn more about how our best-in-class gene editing screening kits and reagents can work to make your gene editing workflows efficient, fast, and simple, from start to finish.
Find the original article on the Takara BioView Blog
Allen, F. et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 37, 64–72 (2019).
Chen, W. et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 47, 7989–8003 (2019).
Shen, M. W. et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018).

If you enjoyed reading our articles, why not sign up to our blog mailing list? You'll get new articles straight to your inbox as they're released!



